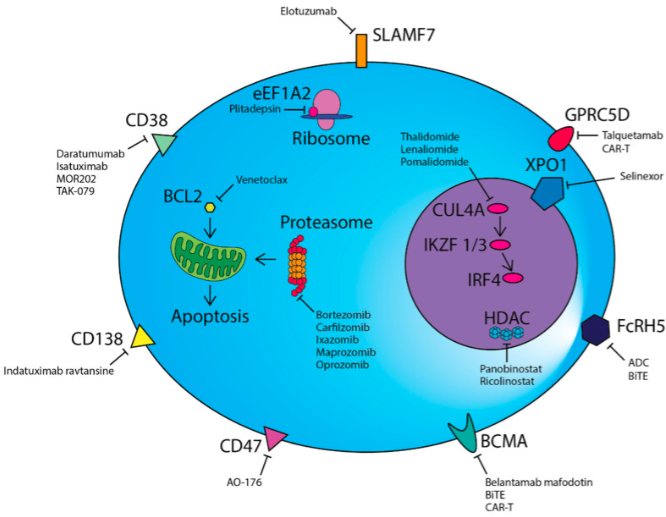
近年来,随着人们对淋巴组织增殖性疾病(LPD)认识地不断深入,一些具有(yǒu)低度恶性潜能(néng)或低度恶性的LPD先后被报道。这些病变比较罕见,转化或进展為(wèi)高级别淋巴瘤的风险很(hěn)低,可(kě)以兼具炎性病变和侵袭性淋巴瘤的某些临床和病理(lǐ)學(xué)特征,因此这类病变的诊治具有(yǒu)很(hěn)大的困难和挑战。这组病变主要包括以下几类:
(1)某些发病机制或临床意义不明的LPD,包括惰性T淋巴母细胞性增生和意义不明的单克隆性γ球蛋白血症等。
(2)某些初诊时处于最初期阶段的淋巴瘤,如"原位淋巴瘤"和单克隆性B淋巴细胞增多(duō)症(MBL)及"组织型MBL"等,前者包括原位滤泡性肿瘤和原位套细胞性肿瘤等;此类疾病肿瘤细胞虽伴有(yǒu)遗传學(xué)异常和单克隆增生,但未侵犯相邻组织结构。
(3)某些交界性LPD/惰性淋巴瘤,除了人们所熟知的无遗传學(xué)异常的"幽门螺杆菌(HP)感染相关性胃部早期黏膜相关淋巴组织结外边缘區(qū)淋巴瘤"和淋巴瘤样丘疹病等外,还包括一些少见类型如十二指肠型滤泡性淋巴瘤(FL)、儿童型滤泡性淋巴瘤(PFL)、儿童结内边缘區(qū)淋巴瘤(PNMZL)等。
(4)某些病毒驱动的LPD,如水疱痘疮样LPD和EB病毒阳性皮肤黏膜溃疡等。
(5)某些特殊类型的T细胞LPD(T-LPD)或NK细胞LPD(NK-LPD),如原发性皮肤CD4阳性的小(xiǎo)/中等T-LPD、原发性皮肤肢端CD8阳性T细胞淋巴瘤、原发头颈區(qū)黏膜或局限于包囊内的乳房假體(tǐ)相关性间变性大细胞淋巴瘤、胃肠道惰性T-LPD、NK细胞淋巴瘤样胃肠病等。对上述病变的深入认识有(yǒu)助于避免误诊和过度临床治疗,因此具有(yǒu)重要意义。
一、惰性B细胞LPD(B-LPD)/淋巴瘤
1.PFL
在2008版WHO分(fēn)类中,PFL被认為(wèi)是一种发生于儿童的滤泡性淋巴瘤的变异型,是FL的暂定分(fēn)类。近年来,逐渐认识到这些病变具有(yǒu)独特的发病人群以及形态學(xué)和生物(wù)學(xué)特征,因此2016版WHO分(fēn)类将以往的"儿童滤泡性淋巴瘤"正式命名為(wèi)"PFL" ,从而成為(wèi)一种独立的病变类型和FL的少见形式。PFL好发于儿童及18~30岁的年轻成人,偶见于老年人。男女患病比例為(wèi)4∶1(普通型FL常见于女性),病变主要位于颈部淋巴结;结外病变部位主要见于睾丸、附睾、咽淋巴环以及胃肠道等。无论是发生于儿童还是发生于成人的PFL,病变均通常较為(wèi)局限,多(duō)為(wèi)Ⅰ期,局部切除后采取观察、等待或小(xiǎo)剂量化疗后预后较好,复发率低,呈惰性临床病程。
PFL形态學(xué)与成人类似,淋巴结结构全部或部分(fēn)破坏,增生滤泡呈膨胀性、地图状或融合片状分(fēn)布,常见"星空"现象,淋巴滤泡极性丢失,大部分(fēn)表现為(wèi)3级FL细胞學(xué)特征,Ki-67阳性指数高(>30%)。免疫表型方面,肿瘤细胞多(duō)呈bcl-2阴性,但也有(yǒu)20%病例呈弱阳性表达;有(yǒu)23%的病例呈CD43阳性表达。分(fēn)子基因學(xué)方面,所有(yǒu)PFL均出现单克隆性Ig基因重排。与普通型FL不同,PFL缺乏t(14;18)(q32;q21)所导致的IgH/bcl-2基因易位重排,也缺乏bcl-6和MYC基因重排。
PFL的确诊需要达到以下标准:显著膨胀的大滤泡,具有(yǒu)Ig基因重排;细胞學(xué)3级,滤泡由中心细胞和中心母细胞样细胞构成;高增殖指数: Ki-67阳性指数>30%;缺乏bcl-2基因重排。日常工作实践中,PFL需要与"FL,3级"相鉴别(尤其是成人病例)。鉴于PFL应该是一种具有(yǒu)低度恶性潜能(néng)的克隆性增生性病变,当FL内出现弥漫性區(qū)域时不能(néng)诊断為(wèi)PFL,尤其是出现局灶性弥漫性大B细胞淋巴瘤(DLBCL)成分(fēn)时更是如此。同时,PFL还需要与患者年龄小(xiǎo)于30岁的普通型FL相鉴别。PFL多(duō)发生于结内,且结内PFL患者绝大部分(fēn)為(wèi)男性,儿童或青少年,中位年龄15岁;病变多(duō)位于头颈部,100%的病例示CD10阳性表达,有(yǒu)18%的病例示bcl-2弱阳性表达但缺乏t(14;18);5.5%病例示MUM1阳性(可(kě)同时CD5阳性)。而年龄小(xiǎo)于30岁的普通型FL患者多(duō)為(wèi)女性,且几乎均為(wèi)年轻成人(中位年龄為(wèi)24岁,均大于18岁);分(fēn)级多(duō)為(wèi)1~2级,3A级相对少见;83%病例表达bcl-2。少部分(fēn)PFL发生于咽淋巴环,此时滤泡内肿瘤细胞示MUM1和bcl-6均呈阳性表达(100%),3/6病例出现IRF4断裂;仅57%病例示CD10阳性,轻链限制性表达罕见,63%病例显示肿瘤性滤泡呈bcl-2阳性表达,但同样无bcl-2/IgH易位[9]。因此,关于"发生于儿童咽淋巴环的FL"是否应归至"PFL" ,目前尚有(yǒu)不同的观点。此外,PFL还需要与儿童结内边缘區(qū)淋巴瘤等相鉴别。
2.伴有(yǒu)IRF4易位的大B细胞淋巴瘤
伴有(yǒu)IRF4易位的大B细胞淋巴瘤是2016 WHO新(xīn)的暂定分(fēn)类,属于成熟B细胞淋巴瘤类型。该病变常见于儿童和年轻成人,多(duō)累及咽淋巴环和/或颈部淋巴结,大部分(fēn)為(wèi)低级别病变。临床多(duō)為(wèi)早期阶段,较PFL侵袭性强,但治疗反应好;个别进展期病变也可(kě)累及腹腔淋巴结和胃肠道。形态學(xué)上,该病变常呈滤泡性、滤泡性和弥漫性或弥漫性分(fēn)布,类似于"FL,3B级"或DLBCL。肿瘤细胞同时呈IRF4/MUM1和bcl-6弥漫强阳性,增殖指数高,半数以上病例表达bcl-2和CD10;当CD10阴性表达时常呈CD5阳性表达,但也有(yǒu)个例显示CD10和CD5同时阳性表达。基因表达谱研究结果显示病变多(duō)為(wèi)生发中心内B细胞来源。分(fēn)子病理(lǐ)學(xué)上,该病变多(duō)有(yǒu)Ig/IRF4基因重排,部分(fēn)合并bcl-6基因重排,但缺乏bcl-2或MYC基因重排;少数无IRF4基因重排,但呈IRF4/MUM1蛋白强阳性表达。由于伴有(yǒu)IRF4易位的大B细胞淋巴瘤和PFL均发生于儿童和青少年、形态學(xué)特征相似且均缺乏t(14;18),因此有(yǒu)文(wén)献将其归為(wèi)淋巴结PFL。
不同的是,伴有(yǒu)IRF4易位的大B细胞淋巴瘤常累及咽淋巴环,且男女患者比例大致相当(未见PFL中男性患者為(wèi)主的特征)、可(kě)出现弥漫性區(qū)域、共表达IRF4/MUM1和bcl-6、常表达bcl-2但缺乏t(14;18)以及与IRF4/IgH基因易位和bcl-6基因异常有(yǒu)关。尽管该肿瘤相较于其他(tā)的儿童型FL具有(yǒu)更强的侵袭性形态學(xué)特征,但经治疗的患者预后极好。
3.十二指肠型滤泡性淋巴瘤
在2008年WHO分(fēn)类中指出"原发胃肠道的FL"是FL的变异型之一,而2016年WHO新(xīn)分(fēn)类则强调十二指肠型FL(DFL)和伴1p36缺失的弥漫性FL(diffuse-appearing FL)具有(yǒu)有(yǒu)别于其他(tā)原发胃肠道FL的独特性质。DFL病变局限于十二指肠黏膜或黏膜下层,与原位滤泡性肿瘤的特点有(yǒu)多(duō)量重叠之处,某些特征还与MALT相似。分(fēn)期低(多(duō)為(wèi)ⅠE或ⅡE),临床上呈惰性病程,预后良好,部分(fēn)患者可(kě)自愈,部分(fēn)患者可(kě)以选择"观察和等待"策略。
4.儿童结内边缘區(qū)淋巴瘤(PNMZL)
PNMZL是2016年WHO分(fēn)类中的新(xīn)增加类型,该肿瘤于1997年由Elenitoba-Johnson等[12]首先报道。好发于儿童和年轻人,中位年龄16岁,亦可(kě)见于成年人(最大年龄66岁)。男性患者為(wèi)主,常為(wèi)无症状、局限性病变(90%病例為(wèi)Ⅰ期),病变多(duō)累及头颈部淋巴结和结外位点。病灶单纯切除后预后良好,复发率低,治疗后生存期長(cháng)。形态學(xué)上,淋巴结正常结构破坏,淋巴滤泡增生、不规则、边缘破坏,边缘區(qū)保存或膨胀扩大呈进行性生发中心转化(PTGC)样改变,滤泡间區(qū)明显扩大,可(kě)见滤泡植入或细胞呈浆样分(fēn)化。PTGC是相对特征性改变,见于66%的PNMZL和10%无症状反应性增生病例。也有(yǒu)文(wén)献提出该类型淋巴瘤伴有(yǒu)"旺炽性淋巴滤泡增生"的概念。免疫表型和分(fēn)子病理(lǐ)特征与成人结内边缘區(qū)淋巴瘤(NMZL)相似,肿瘤细胞表达CD20、CD79a和CD19,伴有(yǒu)PTGC的膨大滤泡结构呈IgD阳性表达,CD23显示树突网;73%病例免疫组织化學(xué)染色或流式细胞术结果显示轻链限制性表达,83%病例异常表达CD43;肿瘤细胞常呈bcl-2阳性但缺乏bcl-2、bcl-6、IgH和MYC基因重排。85%病例显示Ig单克隆性基因重排,21%的病例出现染色體(tǐ)18三體(tǐ)。
PNMZL需要与以下病变相鉴别:
(1)经典型FL伴边缘區(qū)分(fēn)化或PFL:常无PTGC,肿瘤细胞示CD10、bcl-6阳性表达,经典型FL常见bcl-2表达且t(14;18)易位;而PNMZL中残存的生发中心内和滤泡间區(qū)出现多(duō)量PD1阳性的T细胞可(kě)以有(yǒu)助于鉴别PFL;PNMZL的形态學(xué)和免疫组织化學(xué)特点与PFL多(duō)有(yǒu)重叠,但PNMZL缺乏频发的细胞遗传學(xué)异常。
(2)儿童不典型性边缘區(qū)淋巴组织增生:二者临床和形态學(xué)相似,但儿童不典型性边缘區(qū)细胞增生常发生于结外,如扁桃體(tǐ)和阑尾等,呈Ig轻链限制性表达,可(kě)表达CD43;但Ig基因重排阴性,可(kě)以帮助鉴别;儿童结内非典型性边缘區(qū)淋巴组织增生常与流感嗜血杆菌有(yǒu)关。
5.单克隆B淋巴细胞增多(duō)症(MBL)
MBL是指无任何临床症状的健康受试者外周血中发现克隆性增多(duō)的B细胞亚群,数量<5×109/L。随着自动化血细胞计数仪和流式细胞仪的广泛应用(yòng),目前MBL诊断率显著提高,这种实验室检测异常可(kě)见于12%的正常人。MBL的免疫表型存在异质性,根据细胞表面CD5分(fēn)子存在与否分(fēn)為(wèi)3类:慢性淋巴细胞性白血病样MBL(CLL-like MBL)约占75%,呈CD5+但低水平表达CD20;不典型CLL样MBL(atypical CLL-like MBL)呈CD5+且高水平表达CD20;CD5-的MBL(non-CLL MBL)。虽然大部分(fēn)MBL呈CLL样,且MBL几乎发生于所有(yǒu)CLL/SLL之前,但目前并未明确其為(wèi)CLL的前驱病变。在2016年WHO分(fēn)类中,MBL的诊断标准不变,但强调根据克隆性B细胞的数量分(fēn)為(wèi)低计数组和高计数组两组。低计数组是指克隆性B细胞数量<0.5×109/L,该组病变可(kě)能(néng)与老龄化和/或慢性抗原刺激等相关,见于5%的40岁以上人群,进展风险极低、无需常规随访;高计数组则指克隆性B细胞(0.5~5.0)×109/L,该组病变是恶性淋巴肿瘤的一种前驱病变,与Rai 0期CLL(>5×109/L)的表型和遗传學(xué)/分(fēn)子學(xué)特征相似,IgHV突变更常见;年进展率為(wèi)1%~2%,需常规随访。部分(fēn)non-CLL表型的MBL与SMZL密切相关。最近,有(yǒu)研究提出"组织型MBL"的概念,即"SLL"侵犯淋巴结但无"增殖中心" ,淋巴结直径<1.5 cm(基于CT扫描),进展率低。
6.惰性白血病样非结内性套细胞淋巴瘤
套细胞淋巴瘤(MCL)有(yǒu)两种不同的分(fēn)子致病途径:一种表现為(wèi)无或有(yǒu)微小(xiǎo)IgHV突变且SOX11阳性,病变细胞常累及淋巴结和结外部位,形态學(xué)表现為(wèi)经典型MCL或母细胞样/多(duō)形性MCL,临床上呈现出侵袭性的生物(wù)學(xué)行為(wèi);而另一种则表现為(wèi)IgHV突变但SOX11阴性,病变细胞常不累及淋巴结,但侵犯外周血、骨髓和脾脏等,PET-CT示SUV最大值多(duō)小(xiǎo)于6,临床上呈惰性病程,故称之為(wèi)"惰性白血病样非结内性MCL"。后者若出现继发性分(fēn)子遗传學(xué)异常(如TP53异常),则将表现出侵袭性特征。
7.EBV阳性黏膜皮肤溃疡(EBV+ mucocutaneous ulcer)
于2010年由Dojcinov等首次提出,目前已有(yǒu)50余例报道。由于此类病变通常具有(yǒu)自愈潜能(néng),并对传统治疗(放疗和/或化疗)方法反应良好,因此2016年WHO分(fēn)类将其从EBV阳性DLBCL中分(fēn)离出来成為(wèi)一种暂定疾病类型。该病变40%见于老年免疫力降低者(64~101岁,中位年龄81岁),56%见于医源性免疫抑制患者(如移植后患者、炎症性肠病患者、淋巴瘤等肿瘤患者以及服用(yòng)硫唑嘌呤、甲氨蝶呤或环芽孢菌素A的患者等),4%见于免疫缺陷综合征患者。临床上表现為(wèi)不明原因的持续性浅溃疡,大部分(fēn)為(wèi)单发,16%為(wèi)多(duō)发病变;溃疡边界清楚;好发于口咽黏膜,亦可(kě)发生于皮肤(大部分(fēn)位于面部)和胃肠道等部位;常无系统性淋巴结肿大、肝脾肿大或骨髓累及等。形态學(xué)上,病变内可(kě)见多(duō)形性淋巴细胞浸润,包括小(xiǎo)淋巴细胞、免疫母细胞、不典型性的大淋巴细胞、霍奇金细胞样细胞或R-S样大B细胞,混杂有(yǒu)浆细胞、嗜酸性粒细胞和组织细胞等,坏死和血管浸润可(kě)见,病变基底部有(yǒu)小(xiǎo)的T淋巴细胞组成的边缘。
免疫表型上,免疫母细胞强表达CD10、CD30、MUM1、PAX5和OCT-2以及EB病毒编码的小(xiǎo)mRNA(EBER)阳性;CD20、CD45、CD15、CD79a、BOB.1和bcl-6呈强弱不一的阳性表达。浸润其内的T淋巴细胞呈CD4和CD8阳性。此外,T细胞和浆样凋亡细胞均示EBER阳性。值得警惕的是,有(yǒu)38.9%病例可(kě)出现Ig基因重排;69%病例可(kě)出现TCR基因重排。EB病毒阳性黏膜皮肤溃疡患者外周血中检测不到EBV的DNA,可(kě)以有(yǒu)助于鉴别EB病毒阳性移植后LPD。
二、惰性T/NK-LPD/淋巴瘤
1.乳房植入物(wù)相关性间变性大细胞淋巴瘤(BI-ALCL)
BI-ALCL是2016年WHO分(fēn)类中新(xīn)增的间变性淋巴瘤激酶(ALK)-ALCL暂定分(fēn)类,最初于1997年被首次报道。该病变发生率极低,常与隆胸使用(yòng)的硅胶树脂或盐溶液填充物(wù)有(yǒu)关,一般在隆胸后3-19年(中位时间10年)后发生。临床表现為(wèi)乳房肿胀,假體(tǐ)与纤维包囊间出现浆液性液體(tǐ)。形态學(xué)上,病变表现為(wèi)ALK-ALCL特征,肿瘤细胞主要局限于腔内或周围纤维性包膜内,通常不浸润周围组织。肿瘤细胞呈CD30弥漫强阳性表达,细胞毒性标志(zhì)物(wù)阳性,上皮细胞膜抗原(EMA)表达强弱不一,ALK阴性;可(kě)出现克隆性TCR基因重排。临床上,当病变细胞不侵犯包囊时呈惰性经过,切除包囊和假體(tǐ)后预后良好;当病变穿透包囊时,则有(yǒu)淋巴结受侵和全身播散可(kě)能(néng)。
2.原发性皮肤肢端CD8阳性T细胞淋巴瘤(primary cutaneous acral CD8+ T-cell lymphoma)
旧称為(wèi)"皮肤原发性惰性CD8阳性淋巴组织增生" ,2016年WHO分(fēn)类中将其认為(wèi)是一种新(xīn)的暂定分(fēn)类,2007年由Petrella等首先报道。患者中位发病年龄55岁,是一种绝大多(duō)数情况下侵犯耳(偶见于鼻和眼睑)的结节性皮肤损害,具體(tǐ)表现為(wèi)边界不清的红斑、丘疹或结节;病变多(duō)单发,偶可(kě)见病变呈双侧对称分(fēn)布。临床表现呈惰性,虽可(kě)复发或多(duō)灶性发生,但所有(yǒu)行病灶局部切除的患者均存活,显示预后良好。因此,需要注意避免过度治疗。形态學(xué)表现為(wèi)真皮、皮下淋巴样细胞浸润,不累及表皮;瘤细胞形态单一、中等大小(xiǎo),核不规则,呈"母细胞样" ,有(yǒu)时呈印戒细胞样改变;无坏死,无血管侵犯现象。肿瘤细胞呈CD3、CD8和T细胞核内抗原(TIA)1阳性,CD4、CD56和EBER阴性,Ki-67阳性指数低(<15%),可(kě)同时表达CD68。本病必须要与"外周T细胞淋巴瘤,非特指型"相鉴别,以避免过度治疗。
3.原发皮肤CD4阳性小(xiǎo)/中等大小(xiǎo)T细胞增殖性疾病(primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder)
旧称之為(wèi)"原发性皮肤CD4阳性小(xiǎo)/中等大小(xiǎo)T细胞淋巴瘤" ,其可(kě)能(néng)是皮肤对某些不明原因長(cháng)期刺激的局部自限性的克隆反应,而不完全达到诊断恶性的标准,因此2016年WHO分(fēn)类中将其降级為(wèi)LPD以反映其不确定的恶性潜能(néng),但保留為(wèi)一种临时病种。患者中位年龄63岁,头颈部是最常见的受累部位,其次是躯干部。临床行為(wèi)几乎均為(wèi)惰性,恶性潜能(néng)不确定。79%病例单发,8%病例显示蕈样霉菌病(MF)样斑块。形态學(xué)表现為(wèi)真皮内致密淋巴样细胞浸润,呈弥漫分(fēn)布或结节性浸润,可(kě)形成簇状或假菊形团;细胞小(xiǎo)至中等大小(xiǎo);混杂不等量的嗜酸性粒细胞、组织细胞等炎性细胞。免疫表型上,细胞為(wèi)滤泡辅助T细胞(TFH)表型,但不具备结内TFH淋巴瘤的频发突变。细胞表达CD3和CD4,PD1亦常呈阳性表达;CD8、CD56和CD30均呈阴性;分(fēn)别有(yǒu)52%和84%的病例失表达CD5和CD7。另外,肿瘤可(kě)混杂多(duō)量B细胞;EBER检测為(wèi)阴性,TCR克隆性重排阳性。单个病灶者采用(yòng)外科(kē)切除术和/或放疗后,仅有(yǒu)12%病例出现复发;而对于多(duō)发病灶者,主要采取外用(yòng)类固醇、氮芥治疗和光疗法等,但复发率高达69。
4.胃肠道惰性T/NK淋巴细胞增殖性疾病
胃肠道T细胞和NK细胞淋巴瘤常為(wèi)侵袭性疾病,致死率高,但近年来也有(yǒu)部分(fēn)报道显示起源自T或NK细胞的LPD/淋巴瘤具有(yǒu)惰性临床行為(wèi),这些少见病变与炎症性肠病、T或NK细胞淋巴瘤的临床和病理(lǐ)特征可(kě)以非常相似,导致诊断困难。
2016年WHO分(fēn)类将"胃肠道惰性T淋巴细胞增殖性疾病"作為(wèi)一种新(xīn)的暂定分(fēn)类。这是一种来源于T淋巴细胞的罕见疾病,根据肿瘤细胞的免疫表型显示分(fēn)為(wèi)胃肠道惰性CD4+ T-LPD(相对少)与胃肠道惰性CD8+ T-LPD。二者中位年龄相近(51.5岁和45.0岁),男性患者均稍多(duō)于女性患者。
临床症状可(kě)表现為(wèi)腹泻、腹痛腹胀、消瘦等。影像學(xué)上常见肠周围淋巴结肿大。内镜下,前者表现為(wèi)肠道结节、息肉、肠瘘和浅表溃疡,少数黏膜可(kě)无异常;后者常為(wèi)多(duō)灶性累及,好发于小(xiǎo)肠和结肠,可(kě)呈现颗粒状、皱襞增厚、结节或正常外观,常伴肠黏膜水肿、多(duō)量小(xiǎo)息肉或糜烂等。组织學(xué)上,在黏膜固有(yǒu)层或其下方可(kě)见肿瘤细胞呈致密而膨胀的结节状增生,黏膜表面腺體(tǐ)可(kě)被肿瘤细胞挤压、变形、扭曲,但并无淋巴上皮病变;偶尔可(kě)见肿瘤细胞浸润至黏膜肌层和黏膜下层。高倍镜下,膨胀结节内可(kě)见小(xiǎo)到中等大小(xiǎo)、形态略不规则的淋巴样细胞,核型可(kě)不规则,核染色质成熟,核仁不明显,细胞质稀少,偶尔可(kě)见嗜酸性粒细胞与淋巴细胞混合存在。两种疾病的肿瘤细胞的免疫表型均為(wèi)TCR αβ型,CD10、CD30、CD56呈阴性;Ki-67阳性指数约為(wèi)5%~10%,提示该病為(wèi)惰性进展。与胃肠道惰性CD4+ T-LPD不同的是,胃肠道惰性CD8+ T-LPD还可(kě)表达细胞毒性标记(如TIA1、颗粒酶B阳性)。分(fēn)子基因學(xué)显示二者均有(yǒu)TCR β或γ基因重排,提示病变均来源于T细胞的单克隆增殖。
目前尚未明确相关的致病因素,可(kě)能(néng)為(wèi)某一抗原或刺激物(wù)持续刺激并导致单克隆性T细胞群的增生反应。肿瘤细胞示EBER阴性,提示胃肠道惰性T-LPD可(kě)能(néng)与EB病毒感染无关。胃肠道惰性NK-LPD又(yòu)称為(wèi)"淋巴瘤样胃病" 、"NK细胞肠病" ,与食物(wù)抗原或幽门螺旋杆菌感染等有(yǒu)关,其内镜下表现、临床病理(lǐ)學(xué)及影像學(xué)特点等与胃肠道惰性CD4+ T-LPD、CD8+ T-LPD多(duō)有(yǒu)重叠,包括EBER阴性。女性患者略多(duō)于男性患者。患者常表现為(wèi)腹痛不适等(NK细胞肠病)或无症状(淋巴瘤样胃病)。淋巴瘤样胃病多(duō)局限于胃,呈浅表隆起或平坦性病变,病灶直径约1.0 cm,红色、伴或不伴糜烂溃疡。NK细胞肠病多(duō)累及食管、胃和肠多(duō)个位点,病变最大径约1.0~2.0 cm,外观多(duō)样,可(kě)表现為(wèi)充血灶、黏膜结节、牛眼样红斑或浅表出血溃疡。免疫表型上,肿瘤细胞示cCD3、CD7、CD56和细胞毒性标志(zhì)物(wù)TIA1、颗粒酶B、穿孔素阳性,但sCD3、CD5、CD4和B系或髓单核系标志(zhì)物(wù)均阴性,Ki-67阳性指数约10%~30%。淋巴瘤样胃病多(duō)自发消退(平均2.8年,0.3~12.1年),而NK细胞肠病多(duō)持续存在(平均3.0年,1.8~10.0年),患者对化疗不敏感。该病主要与侵袭性NK细胞白血病或NK/T细胞淋巴瘤等相鉴别,后两者细胞异型性更明显,常见坏死,肿瘤细胞Ki-67阳性指数高且EBER阳性。
5.水疱痘疮样淋巴组织增殖性疾病(hydroa vacciniforme-like lymphoproliferative disorder)
T/NK细胞型慢性活动性EB病毒感染(CAEBV)既可(kě)表现為(wèi)局限性、惰性的皮肤病变,也可(kě)表现出系统性症状。前者旧称"水疱痘疮样淋巴瘤" ,好发于南美和亚洲儿童,有(yǒu)进展為(wèi)系统性淋巴瘤的风险,与慢性活动性EB病毒感染相关。临床过程呈多(duō)样性:可(kě)表现為(wèi)蚊子叮咬后过敏反应,或发热、肝脾肿大及淋巴结肿大,伴或不伴皮肤表现等更為(wèi)全身性的病变形式。免疫调节和/或免疫抑制剂治疗后可(kě)获得暂时缓解。因此,2016年WHO分(fēn)类将其降级更名為(wèi)"水疱痘疮样LPD" 。
三、其他(tā)
其他(tā)惰性LPD还包括伴1p36缺失的弥漫性滤泡性淋巴瘤、T淋巴母细胞增生、淋巴瘤样丘疹病、黏膜CD30阳性T-LPD、蕈样肉芽肿和T大颗粒细胞白血病以及年轻人扁桃體(tǐ)DLBCL等。
近年来,有(yǒu)文(wén)献提出"伴1p36缺失的弥漫性FL"的概念,但其目前还未列入独立的疾病类型。该肿瘤多(duō)為(wèi)早期病变(Ann Arbor Ⅰ~Ⅱ期),多(duō)表现為(wèi)局限性、大的腹股沟肿块。形态學(xué)表现為(wèi)弥漫性低级别FL(大部分(fēn)為(wèi)2级),滤泡结构罕见;免疫表型上,肿瘤细胞示CD23阳性表达,CD10和bcl-6表达可(kě)减弱或呈阴性。分(fēn)子遗传學(xué)上,该肿瘤无t(14;18)/bcl-2基因易位重排,96.6%(28/29)病例伴有(yǒu)1p36缺失。基因表达谱显示该肿瘤虽属于经典型FL谱系,但其与经典型FL又(yòu)有(yǒu)明显差异。
T淋巴母细胞增生也是新(xīn)近报道的病变现象,该病变于1999年由Velankar等首先报道,报道中的患者年仅16岁,于异位胸腺瘤中出现异常淋巴母细胞样细胞,且这些细胞表达末端脱氧核苷酸转移酶(TdT)、CD1a、CD3,并共表达CD4和CD8,但未见TCR基因重排。患者行肿块切除后未经辅助治疗,虽出现多(duō)次复发,但预后良好。后来,有(yǒu)研究发现该现象亦可(kě)在肝细胞性肝癌和Castleman病中出现。2013年,Ohgami等提出该病变的诊断标准:
(1)活检标本中出现片状TdT阳性的T细胞;
(2)周围淋巴结构保存且相对正常;
(3)TdT阳性T细胞无明显不典型性;
(4)缺乏胸腺上皮;
(5)TdT阳性T细胞呈多(duō)克隆性;
(6)免疫表型与正常不成熟T淋巴细胞相同;
(7)临床表现出惰性行為(wèi)(随访>6个月无疾病进展)。该病变偶可(kě)表达CD33。
此外,也有(yǒu)文(wén)献报道显示一位51岁中國(guó)女性患者子宫平滑肌瘤内出现惰性B淋巴母细胞增生。同样,目前该病变也不是独立的疾病类型。
黏膜CD30阳性T-LPD病变呈惰性临床经过,与皮肤CD30阳性T-LPD相似,但病例与淋巴瘤样丘疹病无关,细胞不典型性和多(duō)形性明显,肿瘤细胞示CD3、CD4、CD30和细胞毒标记阳性,而CD8和ALK阴性。扁桃體(tǐ)DLBCL的患者如果相对年轻、病程短/发现病变早、形态學(xué)示早期病变、EBER和t(14;18)均阴性且未累及其他(tā)位点等,一般预后也很(hěn)好。
综上,本文(wén)主要介绍了一些近年来新(xīn)认识的惰性LPD,如惰性B-LPD/淋巴瘤以及惰性T或NK-LPD/淋巴瘤等。这些病变在形态學(xué)上有(yǒu)时与相应细胞起源的侵袭性淋巴瘤具有(yǒu)一定相似性。因此,准确识别这类疾病对患者个體(tǐ)化治疗和预后评估等均具有(yǒu)重要意义。准确的临床病理(lǐ)诊断必须整合患者的临床特点、影像學(xué)、组织學(xué)、免疫表型和分(fēn)子遗传學(xué)等信息综合进行。多(duō)种上述病变由于病例量仍较少,目前相应的最佳治疗方案尚不明确,因此需要对患者进行長(cháng)期随访。
上一篇::骨髓增生性疾病如何鉴别?
下一篇::2019 IMW 抢先看 | AlloCAR T™作為(wèi)新(xīn)型靶向BCMA CAR-T疗法,可(kě)用(yòng)于治疗多(duō)发性骨髓瘤

系中國(guó)民(mín)营医疗机构协会理(lǐ)事、石家庄市政协委员、石家庄市劳动模范,从事医疗工作40余年,继承和发扬中國(guó)传统医學(xué)文(wén)化,以血液病的临床研究為(wèi)主题,汲取传统医學(xué)精华,在攻克治疗血液病方面取得了有(yǒu)效成果,积累了丰富 的经验。